Using sophisticated X-ray equipment and highly advanced computational chemistry methods, DTU researchers have shown that it is possible to record ‘molecular movies’ of the interactions between light-activated molecules and their surroundings. This exciting achievement brings us one step closer to exploiting the sun’s energy.
The success of the project is a result a unique partnership between DTU Physics and DTU Chemistry and strong international alliances. The research was conducted in close collaboration with researchers from Stanford University, and supported by partners at European XFEL in Hamburg, Lund University, and KAIST in South Korea.
The researchers were able to follow the detailed interaction between a light-activated photocatalyst and the solvent with which it can react. The new findings, which have just been published in Nature Communications, provide a key step towards a general understanding of the dynamics involved in artificial photosynthesis through which molecules absorb and store solar energy in chemical bonds. Such results are expected to help pave the way for designing light-activated molecules that can produce e.g. hydrogen fuel from water, imitating natural photosynthesis through which nature transforms solar energy and carbon dioxide to plant sugars.
“Photocatalytic systems offer huge potential. If we can understand the underlying dynamics of chemical reaction in light absorbent molecules, we can make rational decisions for how to improve their ability to produce fuels or other important molecules.” says Professor Martin Meedom Nielsen from DTU Physics, who headed the experiments.
Many important dynamics following light-activation of photocatalytic molecules happen on ultra-fast time scales – as fast as few hundred femtoseconds. A hundred femtoseconds or 0.0000000000001 seconds is the time it takes light to travel 0.03 mm – the width of a human hair. This means that it takes highly specialized, sophisticated equipment to capture these dynamics. Martin Meedom Nielsen and his group used a so-called free-electron laser (LCLS in Stanford, California, USA) which they have previously used to track the pathways of electron transport through large molecules. For the present study, the experimental results were extremely hard to interpret, and it was only through the inclusion of computational chemistry results, provided by the group of Professor Klaus Braagaard Møller at DTU Chemistry, that the dynamics could be understood.
“It was very much a ‘eureka moment’ when we found out that the computational chemistry perfectly described the part of the data we had problems understanding. We were all surprised at just how well the computational results agreed with the experimental data” Says Kasper Skov Kjær, a post-doc at DTU Physics, who has analysed the data in collaboration with Tim Brandt van Driel, former post doc at DTU Physics, and now working at LCLS.
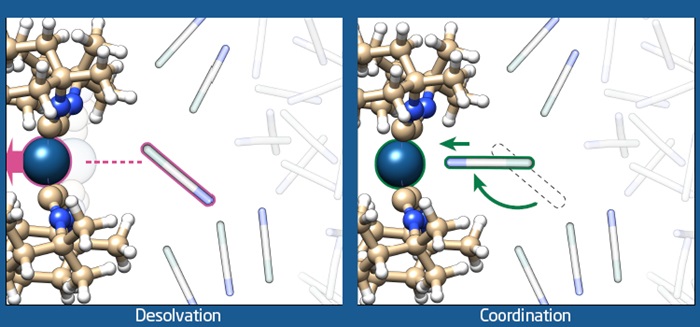
The photocatalyst investigated in these studies is an organometallic compound which represents a prototype of a large class of molecules that perform a whole range of photochemical reactions. The combination of experiments and computational chemistry revealed that light-excitation of the molecule initiates large-scale structural dynamics, and completely changes the nature of the interaction with the surrounding solvent molecules. Before light absorption, the solvent molecules are attracted to the photocatalyst through diffuse and non-specific interaction, the light absorption changes these interactions to very specific interactions causing the solvent molecules to rotate 180 degrees and ‘attach’ to the active site of the photocatalyst. This type of dynamics has never been visualized before.
“I think it is one of the most interesting computational studies we have ever conducted. It shows that our computational chemistry has reached a level of accuracy and complexity where we can predict how a given photocatalytic system will interact with its surroundings following light absorption. Such predictive power of computational chemistry is the most important step towards establishing a situation where we can simulate the behavior of molecules before we start the very complex and time-consuming process of synthesizing them.” says Professor Klaus Braagaard Møller, who headed the computational studies that proved invaluable in interpreting and modeling the recorded data.
“We are looking very much forward to taking the next step in the exploration of molecular dynamics” says Professor Martin Meedom Nielsen, whose group is preparing to take full advantage of the European X-ray Free-Electron Laser which is due to begin operation in Hamburg next year.
Contacts
Martin Meedom Nielsen
Professor
DTU Physics
Phone: +45 45 25 32 26
E-Mail: mmee@fysik.dtu.dk
Klaus Braagaard Møller
Professor
DTU Chemistry
Phone: +45 45 25 24 61
E-Mail: kbmo@kemi.dtu.dk
Source
Danmarks Tekniske Universitet, press release, 2016-11-28.
Supplier
European XFEL
Korea Advanced Institute of Science and Technology (KAIST)
Lund University (Sweden)
nature (Journal)
Stanford University
Technical University of Denmark – DTU
Share
Renewable Carbon News – Daily Newsletter
Subscribe to our daily email newsletter – the world's leading newsletter on renewable materials and chemicals








