Ein Durchbruch in der Kristallographie ermöglicht Forschern den Zugang zu den Bauplänen von tausenden medizinisch und biologisch bedeutenden Biomolekülen. Die neue Methode, die von einem Team unter Leitung von DESY-Wissenschaftler Professor Henry Chapman vom Hamburger Center for Free-Electron Laser Science CFEL entwickelt wurde, eröffnet einen vergleichsweise einfachen Weg, die räumlichen Strukturen von Proteinen und anderen Biomolekülen zu bestimmen, die über bisherige Verfahren in vielen Fällen nicht zugänglich waren. „Unsere Entdeckung erlaubt uns, atomare Details von großen Proteinkomplexen genau abzubilden“, erläutert Chapman, der auch Professor an der Universität Hamburg und Mitglied des Hamburg Centre for Ultrafast Imaging CUI ist. Sein Team stellt die grundlegende Entdeckung in der aktuellen Ausgabe des Fachjournals „Nature“ vor.
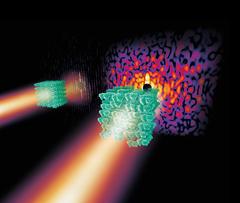
Die räumliche Struktur eines Biomoleküls liefert wichtige Informationen über seine Funktionsweise und kann damit beispielsweise als Basis zur Entwicklung eines Medikaments dienen. Um diese Struktur zu entziffern, nutzen Forscher vor allem die Technik der Kristallographie. Viele der komplexen Biomoleküle ließen sich mit bisherigen Verfahren jedoch praktisch nicht analysieren. Die neue Methode kann mit weniger geordneten Kristallen arbeiten und kommt ohne die sonst benötigten Zusatzinformationen und chemisches Vorwissen aus. „Diese Entdeckung besitzt das Potenzial, eine echte Revolution in der Kristallographie komplexer Materie zu werden“, betont der Vorsitzende des DESY-Direktoriums, Professor Helmut Dosch.
Mit Hilfe der Kristallographie lässt sich die räumliche Struktur eines Kristalls sowie seiner Bestandteile bestimmen, indem der Kristall mit Röntgenstrahlen beleuchtet wird. Die Röntgenstrahlen werden vom Kristall gestreut und erzeugen dadurch ein charakteristisches Muster aus hellen Punkten, sogenannten Bragg-Peaks (benannt nach den britischen Kristallographie-Pionieren William Henry und William Lawrence Bragg). Die Position und Intensität dieser hellen Punkte im Röntgenstreubild liefern Informationen über die Struktur des Kristalls und seiner Bestandteile. Auf diese Weise haben Forscher bereits die atomare Struktur von zehntausenden Proteinen und anderen Biomolekülen bestimmt, die zuvor allerdings aufwändig kristallisiert werden mussten.
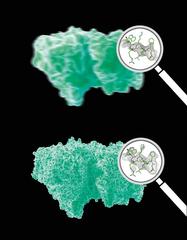
Bei der Kristallographie von komplexen Biomolekülen gibt es jedoch Hürden, durch die eine Strukturbestimmung extrem schwierig oder sogar unmöglich werden kann. Zum einen sind qualitativ hochwertige, besonders regelmäßige Kristalle nötig. Je stärker eine Probe vom perfekten Kristall abweicht, desto weniger Bragg-Peaks sind sichtbar. Dadurch lässt sich die Struktur oft gar nicht mehr bestimmen oder nur ein verschwommenes Abbild des Moleküls mit niedriger Auflösung erzeugen. Die meisten Biomoleküle bilden natürlicherweise keine Kristalle, und es erfordert oft großes Geschick sowie etwas Glück, hochwertige Kristalle aus ihnen zu züchten. Das gilt besonders für die Klasse der Membranproteine, die bei zahlreichen biologischen Prozessen eine wichtige Rolle spielen und auf die rund die Hälfte aller Medikamente zielen.
„Extrem-Sudoku in drei Dimensionen“
Doch selbst mit einem perfekten Kristall lässt sich eine völlig unbekannte Proteinstruktur nicht allein aus den Bragg-Peaks bestimmen. „Diese Aufgabe ist wie Extrem-Sudoku in drei Dimensionen mit Millionen Kästchen aber nur der Hälfte der nötigen Tipps“, erläutert Chapman. In der Kristallographie wird dieses komplizierte Puzzle als Phasenproblem bezeichnet. Der Begriff beschreibt die Tatsache, dass die Phasen der gestreuten Lichtwellen bekannt sein müssen, um die Struktur des Moleküls zu berechnen. Die Phase einer Lichtwelle gibt an, wie sehr ihr Wellenberg dem einer anderen Welle vorauseilt oder hinterherläuft. Die Phasen der einzelnen Wellen lassen sich jedoch nicht messen. Um das Rätsel zu knacken, sind daher weitere Hinweise nötig. Diese lassen sich unter Umständen aus der bereits bekannten Struktur eines chemisch eng verwandten Moleküls gewinnen oder aus dem Vergleich mit Streubildern von Kristallen chemisch leicht veränderter Moleküle. Auch diese Hürde erschwert insbesondere bei großen Molekülkomplexen wie etwa Membranproteinen die Strukturbestimmung.
Chapman entdeckte, dass das Phasenproblem und das Problem der nicht perfekten Kristalle miteinander verbunden sind. Der Schlüssel liegt in einem schwachen, kontinuierlichen Streubild, das bei „unordentlichen“ Kristallen entsteht. Dieses kontinuierliche Streubild gilt in der Regel als störender „Hintergrund“. Daraus lassen sich zwar Einblicke in die Vibrationen und andere Dynamiken der Moleküle gewinnen, für die Strukturanalyse wird es jedoch normalerweise nicht berücksichtigt. Doch wenn die Unordnung im Kristall einzig daher rührt, dass die einzelnen Moleküle leicht von ihrer Idealposition im Kristall verschoben sind, bekommt der vermeintlich störende „Hintergrund“ einen sehr viel komplexeren Charakter: Er enthält das komplette kontinuierliche Streubild der Einzelmoleküle im Kristall.
„Würde man ein einzelnes Molekül mit Röntgenstrahlen beleuchten, würde es ein kontinuierliches Streubild ohne irgendeinen Bragg-Peak erzeugen“, erläutert Erstautor Dr. Kartik Ayyer aus Chapmans CFEL-Gruppe. „Das Muster wäre allerdings extrem schwach und sehr schwer zu messen. Aber der ‘Hintergrund’ in unserer Analyse ist wie eine Aufsummierung zahlreicher Einzelaufnahmen individueller Moleküle. Wir benutzen den Kristall quasi nur, um eine Vielzahl gleich ausgerichteter Moleküle gemeinsam in den Strahl zu befördern.“ Das kontinuierliche Streubild liefert ausreichend Informationen, um das Phasenproblem direkt zu lösen, ohne dass irgendetwas über das untersuchte Molekül bekannt sein muss. In Analogie zum Sudoku-Puzzle ergeben die Messungen nun genug Hinweise, um stets die richtige Antwort zu finden.
Die besten Kristalle sind nicht perfekte Kristalle
Dieses Konzept führt zu einem Paradigmenwechsel in der Kristallographie: Die am besten geordneten Kristalle sind bei dem neuen Verfahren nicht mehr die besten für die Analyse. Am besten eignen sich leicht ungeordnete Kristalle. „Erstmals haben wir Zugang zu Streubildern einzelner Moleküle – das gab es zuvor in der Kristallographie noch nie. Dabei wissen wir seit langem, wie sich das Streubild einzelner Moleküle analysieren lässt, wenn man es denn messen kann.“ Die Technik der sogenannten kohärenten Röntgenbeugung mit Hilfe von Freie-Elektronen-Lasern hat hierzu sehr leistungsfähige Algorithmen geliefert. „Man muss nicht einmal die Chemie kennen“, betont Chapman. „Aber man kann sie direkt aus den dreidimensionalen Bildern erkennen, die man bekommt.“
Um ihre neue Technik experimentell zu testen, tat sich Chapmans Gruppe mit dem Team von Professor Petra Fromme und weiteren Forschern von der Arizona State University, der Universität von Wisconsin, der griechischen Stiftung für Forschung und Technology – Hellas FORTH sowie dem US-Beschleunigerzentrum SLAC zusammen. Die Wissenschaftler nutzten den weltstärksten Röntgenlaser LCLS am SLAC, um „unordentliche“ Kristalle eines Membranproteinkomplexes namens Photosystem II zu untersuchen, der Teil der Photosynthesemaschinerie in grünen Pflanzen ist.
Die Analyse des kontinuierlichen Streubilds verbesserte in dem Versuch die Detailgenauigkeit gegenüber der reinen Auswertung der Bragg-Peaks unmittelbar um etwa ein Viertel von 4,5 Ångström auf 3,5 Ångström. Ein Ångström ist ein Zehntel Nanometer (milliardstel Meter) und entspricht in etwa dem Durchmesser eines Wasserstoffatoms. Das resultierende Bild zeigt dadurch Details des Moleküls, die sonst nur durch die rechnerische Anpassung an ein chemisches Modell sichtbar werden. „Das ist eine ziemlich großer Schritt bei der Untersuchung von Biomolekülen“, betont Ko-Autor Dr. Anton Barty von DESY. „Und wir können die räumliche Auflösung weiter verbessern, wenn wir mehr Bilder aufnehmen.“ Das Team hatte für diese ersten Versuche nur ein paar Stunden Messzeit zur Verfügung, während eine normale Messkampagne oft einige Tage dauert.
Die Wissenschaftler hoffen nun, noch besser aufgelöste Bilder vom Photosystem II und vielen anderen Makromolekülen mit ihrer neuen Technik gewinnen zu können. „Diese Form der kontinuierlichen Röntgenbeugung hat man tatsächlich schon seit langem bei vielen schlecht streuenden Kristallen beobachtet“, erläutert Chapman. „Man hatte allerdings noch nicht verstanden, dass sich daraus Strukturinformationen gewinnen lassen, daher wurde sie bei der Analyse gewöhnlich unterdrückt. Wir werden jetzt viel damit zu tun haben zu prüfen, ob wir aus alten, ursprünglich verworfenen Daten weitere Molekülstrukturen gewinnen können.“
Originalarbeit:
Macromolecular diffractive imaging using imperfect crystals; Kartik Ayyer et al.; Nature (2016); DOI: 10.1038/nature16949
Source
Deutsches Elektronen-Synchroton, 2016-02-11.
Supplier
Arizona State University
Deutsches Elektronen-Synchrotron (DESY)
nature (Journal)
SLAC National Accelerator Laboratory
Universität Hamburg
University of Wisconsin, Madison
Share
Renewable Carbon News – Daily Newsletter
Subscribe to our daily email newsletter – the world's leading newsletter on renewable materials and chemicals







